
Při primární (vrozené) imunodeficienci (PID) se v těle nevytvářejí v dostatečné míře některé součásti imunitního systému (imunoglobuliny – Ig, T-lymfocyty, fagocyty, složky komplementu) nebo některé z nich nefungují správně. Jedná se o více než 400 chronických poruch, vznikajících v důsledku genetické vady nebo změn v lidské DNA, v jejichž důsledku může u postiženého jedince existovat zvýšená náchylnost k bakteriálním a virovým infekcím (i atypickými agens – oportunními patogeny) a také vyšší nemocnost (banálními infekcemi i pneumoniemi, sinusitidami, meningitidami a abscesy), přičemž infekce většinou špatně odpovídá na konvenční léčbu. V Evropě trpí PID cca 10 000 osob a v převážné míře se manifestuje jako deficience protilátková, případně jako deficit T-buněk (a kombinované poruchy). Tato patologie se u jedince může objevit, aniž by se PID vyskytovala v rodinné anamnéze.
Pacientovi s imunodeficitem je nutno v rámci substituční imunoglobulinové terapie podávat infuzí nebo injekčně (intravenózně či subkutánně) protilátky získané z krve zdravých jedinců. Další možností léčby je transplantace kostní dřeně či krvetvorných buněk a genová terapie.
Sekundární (získané) imunodeficience (SID) nejsou až tak vzácné jako PID. Nevznikají na podkladě genetické poruchy, nýbrž podvýživy, jiné nemoci (chronická lymfatická leukémie – CLL, mnohočetný myelom – MM) nebo intervence, dysregulací imunitního systému při napadení tkáně hostitelského organismu, často prostřednictvím složitých mechanismů (většinou u neuromuskulárního postižení), a jsou známy i jako polékové reakce (při systémové kortikoterapii, imunosupresivní léčbě a protinádorové chemoterapii či radioterapii). Iatrogenní imunosuprese se také zaznamenává v souvislosti s některými pooperačními stavy (u popálenin nebo po splenektomii). Nejčastějším projevem sekundárních imunodeficiencí jsou rekurentní infekce v oblasti dýchacích cest a trávicího traktu. Při klinických projevech hypogamaglobulinemie má léčba charakter substituční terapie imunoglobuliny.
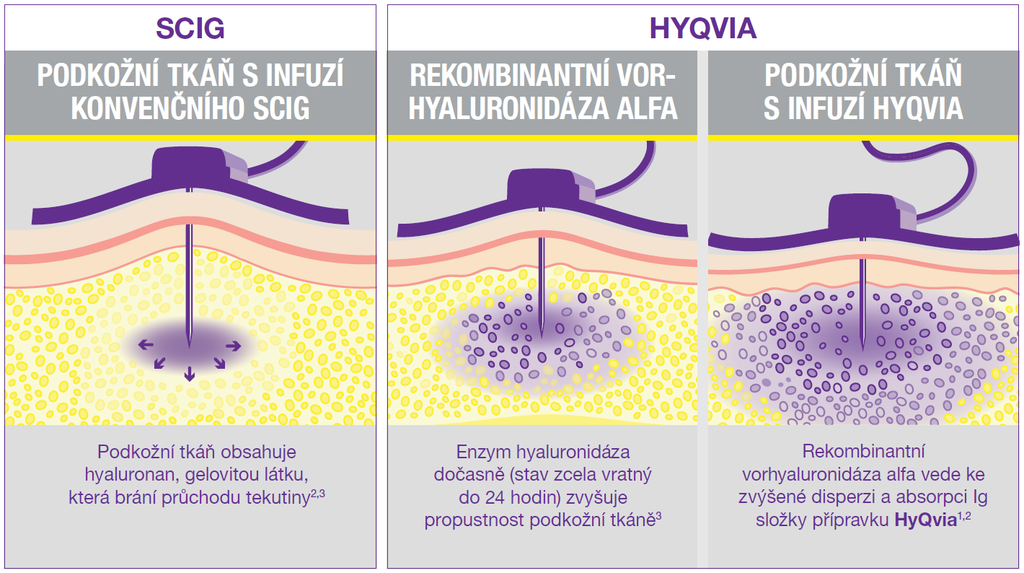
Vedle nitrožilního (i.v.) podání imunoglobulinů (IVIg) se používají také subkutánní (s.c.) aplikace (SCIg). K dispozici jsou i sofistikované produkty (facilitované přípravky – fSCIg), využívající dočasného zvýšení přirozeného obratu hyaluronanu v kůži s otevřením mikroskopických kanálků v subkutánní tkáni, což zvyšuje její propustnost.1 Podkožní podání také zvyšuje komfort léčby – současným trendem je rychlý přesun pacienta do domácího léčení a po zaškolení i domácí podávání Ig.
V této oblasti jsou Vám k dispozici tyto produkty společnosti Takeda:
- HyQvia
- Cuvitru
- Kiovig
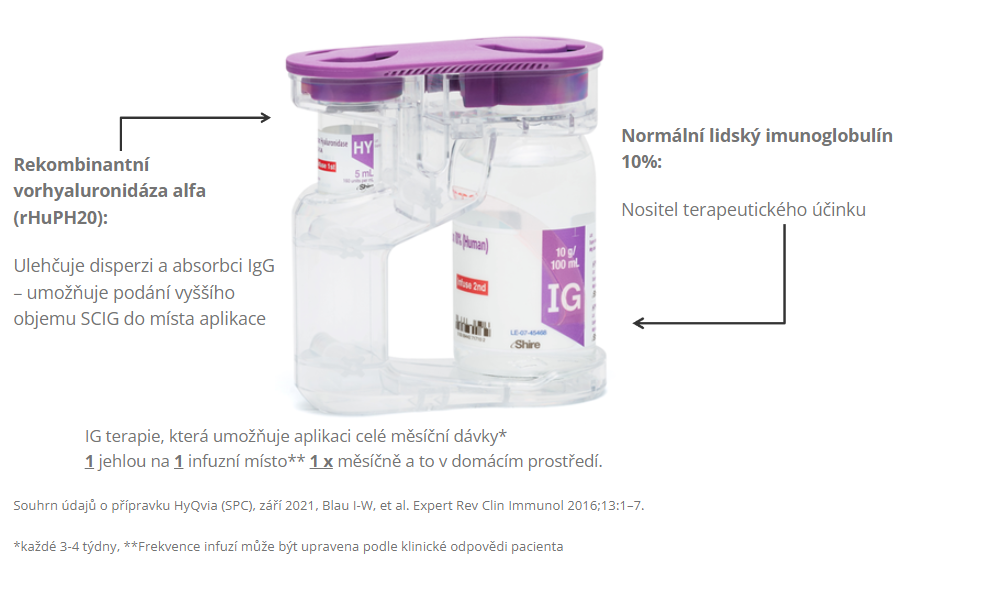
HyQvia je první a jediný SCIg s rekombinantní lidskou hyaluronidázou, která po podkožním podání zvyšuje rozptyl a vstřebávání imunoglobulinů. Má charakter facilitovaného 10% SCIg k substituční terapii dospělých, dětí i dospívajících (0–18 let), nevyžaduje žilní přístup a umožňuje podat najednou celou měsíční dávku Ig (obvykle na jednom infuzním místě). Má 93% biologickou dostupnost a režim podávání lze upravit dle léčebných potřeb pacienta. Příznivým faktem je, že při této léčbě se objevuje méně systémových nežádoucích účinků než u IVIg (8 % vs. 25 %), méně nežádoucích účinků za měsíc než u konvenčních SCIg produktů a že bylo zaznamenáno 2,97 infekcí na pacienta/rok oproti 4,51 při terapii IVIg a jen 0,025 akutních závažných bakteriálních infekcí za pacientorok. Většina (98,7 %) nežádoucích účinků byla považována za mírné nebo středně závažné.2
- Bookbinder LH, Hofer A, Haller MF, et al. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J Control Release 2006;114:230–241.
- Wasserman RL, Melamed I, Stein MR, et al. Recombinant human hyaluronidase-facilitated subcutaneous infusion of human immunoglobulins for primary immunodeficiency. J Allergy Clin Immunol 2012; 130:951–957.e11.
- Souhrn údajů přípravku HyQvia, září 2021.
- Wasserman R. L. et al. J Allergy Clin Immunol. 2012; 130 (4): 951–957.
- Bookbinder L. H. et al. J Control Release. 2006; 114 (2): 230–241.
HyQvia – facilitovaný podkožně podávaný imunoglobulin (fSCIG)
Cuvitru je normální lidský imunoglobulin (SCIg) 20% s podáváním 1× týdně, který někteří pacienti preferují. Francouzská studie ukázala, že co se týká interference při léčbě, jsou pacienti s produkty pro domácí aplikaci SCIg výrazně spokojenější, než je tomu u IVIg, a jsou i spokojenější s SCIg produkty oproti substituci IVIg podávané v nemocnici.1 Potěšitelné je, že u terapie Cuvitru 2 ze 3 pacientů nevykazují lokální nežádoucí účinky a 99,8 % hodnocených infuzí bylo dokončeno bez přerušení, snížení rychlosti nebo ukončení z důvodu nesnášenlivosti.2,3 Výhodou je i to, že Cuvitru umožňuje podání do < 1 hodiny (až 60 ml/h/místo).
- Bienvenu B, Cozon G, Hoarau C, et al. Does the route of immunoglobin replacement therapy impact cohort “Visages“. Orphanet J Rare Dis 2016;11:83.
- Borte M, Kriván G, Derfalvi B, et al. Efficacy, safety, tolerability and pharmacokinetics of a novel human immune globulin subcutaneous, 20%: a phase 2/3 study in Europe in patients with primary immunodeficiencies. Clin Exp Immunol 2017;187:146–159.
- Suez D, Stein M, Gupta S, et al. Efficacy, safety, and pharmacokinetics of a novel human immune globulin subcutaneous, 20 % in patients with primary immunodeficiency diseases in North America. J Clin Immunol 2016;36:700–712.
- Souhrn údajů o přípravku Cuvitru (SPC), září 2021.
Kiovig je normální lidský imunoglobulin 10% k infuznímu podání. Je určen k substituční a imunomodulační terapii dospělých, dětí i dospívajících (0–18 let), především u syndromů primárního imunodeficitu s poruchou tvorby protilátek, sekundárních imunodeficitů u pacientů trpících závažnými nebo opakovanými infekcemi a také například u pacientů s Kawasakiho chorobou, chronickou zánětlivou demyelinizační polyradikuloneuropatií (CIDP), multifokální motorickou neuropatií (MMN) nebo s Guillain-Barrého syndromem. Jeho výhodou je 100% biologická dostupnost.1
- Souhrn údajů o přípravku Kiovig (SPC), květen 2020.
Imunoglobuliny – specifika typů terapie
|
|
|
|
|
| Intravenózní IG (IVIG) | Konvenční SCIG (cSCIG) | Facilitovaný SCIG (fSCIG) | |
|---|---|---|---|
|
Samostatné podání1,2,5 |
|
|
|
|
Typický počet |
|
|
|
|
Objem na infuzi1,6 |
|
|
|
|
Frekvence infuzí1,6 |
|
|
|
|
Biologická dostupnost7 |
|
|
|
|
Celková doba podávání infuze za měsíc3,4,6 |
|
|
|
- Jolles S, et al. Clin Exp Immunol. 2015;179(2):146-160.
- Jolles S. Immunotargets Ther. 2013;2:125-133.
- Kirmse J. Home Healthc Nurse. 2009;27(2):104-111.
- Skoda-Smith S, et al. Ther Clin Risk Manag. 2010;6:1-10.
- Souhrn údajů přípravku HyQvia, září 2021
- Wasserman RL, et al. J Allergy Clin Immunol. 2012;130(4):951-957.
- Wasserman RL. Immunotherapy. 2014.
- Borte M, Kriván G, Derfalvi B, et al. Efficacy, safety, tolerability and pharmacokinetics of a novel human immune globulin subcutaneous, 20%: a phase 2/3 study in Europe in patients with primary immunodeficiencies. Clin Exp Immunol 2017;187:146–159.
C-APROM/CZ/CUVI/0019 Srpen 2022