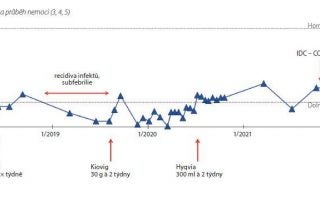
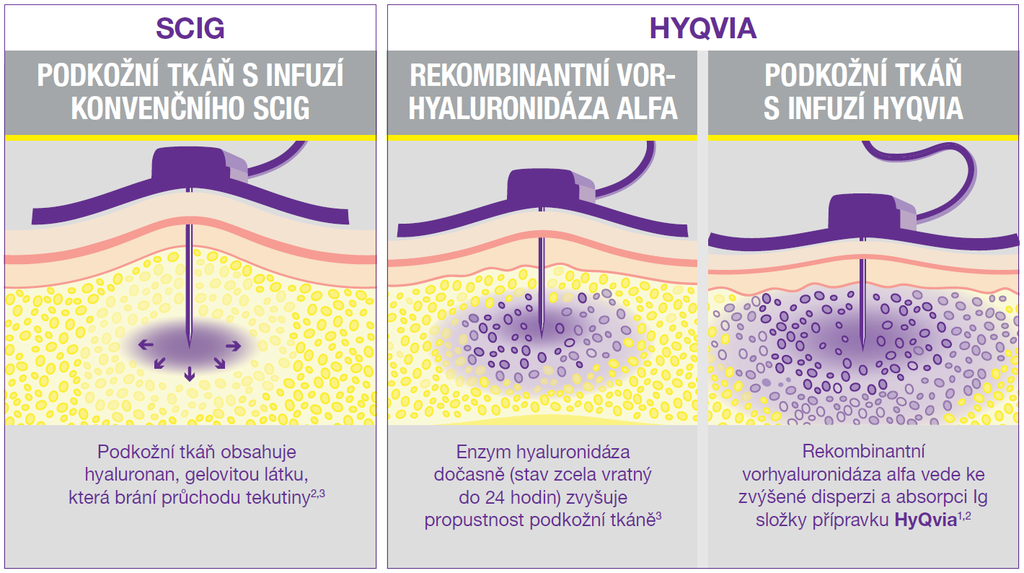
Zkrácené informace o léčivém přípravku
GAMMAGARD S/D, 5 g (10 g) prášek a rozpouštědlo pro infuzní roztok
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Léčivá látka: Immunoglobulinum humanum normale ad usum intravenosum (IVIg). Pomocné látky se známým účinkem: sodík. Jedna injekční lahvička obsahuje 5 g nebo 10 g immunoglobulinum humanum normale ad usum intravenosum (IgG). Maximální obsah imunoglobulinu A (IgA): 3 mg /ml v 5% roztoku.
Indikace: Substituční léčba u dospělých a dětí a adolescentů (0-18 let) u: syndromů primárního imunodeficitu (PID) s poruchou tvorby protilátek; hypogamaglobulinémie a rekurentních bakteriálních infekcí u pacientů s chronickou lymfatickou leukemii, u nichž selhala antibiotická profylaxe; hypogamaglobulinémie a rekurentních bakteriálních infekcí u pacientů s mnohočetným myelomem ve fázi plateau, kteří nereagovali na imunizaci proti pneumokokům; hypogamaglobulinémie u pacientů po alogenní transplantaci hematopoetických kmenových buněk; vrozeného AIDS s rekurentními bakteriálními infekcemi. Nezralé děti s nízkou porodní hmotností. Imunomodulace u dospělých a dětí a adolescentů (0-18 let) u: primární imunitní trombocytopenie (Idiopatická trombocytopenická purpura (ITP)), u pacientů, u nichž je vysoké riziko krvácení, nebo před chirurgickým výkonem k úpravě počtu trombocytů; Guillain-Barrého syndromu; Kawasakiho choroby.
Dávkování a způsob podání: Dávka a režim dávkování závisí na indikaci. Při substituční léčbě je třeba přizpůsobit dávkování individuálně u každého pacienta podle jeho farmakokinetické a klinické odpovědi. Následující režimy dávkování slouží jako vodítko.
Substituční léčba u syndromů primárního imunodeficitu: Režim dávkování by měl být takový, aby nejnižší hladina IgG (měřená před další infuzí) byla minimálně 5-6 g/l. Dosažení rovnováhy trvá tři až šest měsíců od zahájení léčby. Doporučovaná úvodní dávka je 0,4-0,8 g/kg tělesné hmotnosti (TH), po níž následuje dávka minimálně 0,2 g/kg TH podávaná každé tři až čtyři týdny. Dávka potřebná k dosažení minimálních koncentrací 5-6 g/l před další infuzí se pohybuje mezi 0,2-0,8 g/kg/TH/měsíc. Po dosažení rovnovážného stavu se interval mezi dávkami pohybuje od tří do čtyř týdnů. Je třeba měřit hladiny před další infuzí a porovnávat je s incidencí infekcí. Ke snížení počtu infekcí může být zapotřebí zvýšit dávkování s cílem dosáhnout vyšších předinfuzních hladin.
Hypogamaglobulinémie a rekurentní bakteriální infekce u pacientů s chronickou lymfatickou leukémií, u nichž selhala antibiotická profylaxe, hypogamaglobulinémie a rekurentní bakteriální infekce u pacientů s mnohočetným myelomem ve fázi plateau, kteří nereagovali na imunizaci proti pneumokokům, děti s vrozeným AIDS a rekurentními bakteriálními infekcemi: Doporučená dávka je 0,2-0,4 g/kg TH každé tři až čtyři týdny.
Nezralé děti s nízkou porodní hmotností: K profylaxi pozdního rozvoje infekce u nezralých dětí s nízkou porodní hmotností dostávají novorozenci mladší než 7 dnů 0,5 g/kg TH a stejnou dávku o týden později. Poté následuje celkem 5 infuzí každých 14 dnů nebo do propuštění z nemocnice.
Hypogamaglobulinémie u pacientů po alogenní transplantaci hematopoetických kmenových buněk: U léčby infekcí a profylaxe reakce štěpu proti hostiteli se dávky stanovují individuálně. Doporučená dávka je 0,2-0,4 g/kg TH každé tři až čtyři týdny. Předinfuzní hladiny by měly být udržovány nad 5 g/l.
Primární imunitní trombocytopenie: Existují dvě alternativní schémata léčby: 0,8-1 g/kg TH podáno v den jedna; tuto dávku lze opakovat jednou během 3 dnů nebo 0,4 g/kg podáváno denně po dobu dvou až pěti dnů. V případě relapsu je možné léčbu zopakovat.
Guillainův-Barrého syndrom: 0,4 g/kg/den po dobu 5 dnů
Kawasakiho choroba: 1,6-2,0 g/kg TH má být podáno v rozdělených dávkách po dobu dvou až pěti dnů nebo 2,0 g/kg jako jedna dávka. Pacientům by měla být souběžně podávána kyselina acetylsalicylová.
GAMMAGARD S/D 5% (50 mg/ml) má být podáván intravenózně úvodní rychlostí 0,5 ml/kg TH/hod. Obecně se doporučuje pomalejší zahájení infuze u pacientů, kteří začínají s léčbou přípravkem GAMMAGARD S/D nebo jsou převáděni z jiného intravenózního imunoglobulinu. Pokud dobře snášeli několik infuzí střední rychlosti, lze zrychlit až na maximální infuzní rychlost 8 ml/kg TH/hod.
Kontraindikace: Hypersenzitivita na nebo známá přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku.
Upozornění:* Tento léčivý přípravek obsahuje jako pomocnou látku 20 mg glukózy v jednom ml (400 mg/g IgG). To je třeba vzít v úvahu v případě latentního diabetu (kde se může objevit přechodná glykosurie), u diabetu nebo u pacientů na dietě s nízkým obsahem cukru. Některé závažné nežádoucí účinky mohou souviset s rychlostí infuze. Podávání intravenózních imunoglobulinů vyžaduje u všech pacientů: adekvátní hydrataci před zahájením infuze intravenózního imunoglobulinu, sledování výdeje moči, sledování hladin kreatininu v séru, vyhnout se současnému podávání kličkových diuretik. Vzácně může normální lidský imunoglobulin navodit anafylaktický šok s poklesem krevního tlaku, dokonce i u pacientů, kteří předchozí léčbu normálním lidským imunoglobulinem snášeli dobře. Bylo klinicky prokázáno, že existuje souvislost mezi aplikací IVIg a tromboembolickými příhodami, jako je infarkt myokardu, cévní mozková příhoda, plicní embolie a hluboké žilní trombózy. U pacientů s rizikem akutního renálního selhání by měly být přípravky IVIg podávány minimální infuzní rychlostí v nejnižší možné dávce. U pacientů, kterým byly podávány IVIg, byly hlášeny případy nekardiogenního plicního edému. Ve spojení s léčbou IVIg byl hlášen syndrom aseptické meningitidy. Přípravek GAMMAGARD S/D obsahuje protilátky proti krevním skupinám, které mohou působit hemolyticky a navodit potažení erytrocytů imunoglobulinem in vivo, což může vyvolat pozitivní přímou antiglobulinovou reakci (Coombsův test). Po aplikaci imunoglobulinu může v krvi pacienta dojít k přechodnému vzestupu pasivně přenesených protilátek, a tím ke vzniku zavádějících pozitivních výsledků v sérologických testech např. hepatitida A, hepatitida B, spalničky a varicela. Tento léčivý přípravek obsahuje 668 mg sodíku v jedné injekční lahvičce (10 g).
GAMMAGARD S/D se vyrábí z lidské plazmy. Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a zavedení účinných výrobních kroků k inaktivaci / odstranění virů. Přes všechna tato opatření při přípravě léků vyráběných z lidské krve nebo plazmy nelze možnost přenosu infekčních agens zcela vyloučit.
U pacientů léčených IVIg se může objevit hyperproteinémie a zvýšená viskozita séra. Při každé aplikaci přípravku GAMMAGARD S/D důrazně doporučujeme zaznamenat název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Interakce: Podání imunoglobulinu může narušit po dobu minimálně 6 týdnů až 3 měsíců účinnost vakcín, které obsahují živé atenuované viry, např. spalniček, zarděnek, příušnic a planých neštovic. Před očkováním vakcínou, která obsahuje živé atenuované viry, by od podání tohoto přípravku měly uplynout 3 měsíce. V případě vakcíny proti spalničkám může toto narušení přetrvávat až 1 rok. U pacientů očkovaných proti spalničkám by se proto měla zkontrolovat hladina jejich protilátek.
Nežádoucí účinky: Při intravenózním podání normálních imunoglobulinů se mohou výjimečně objevit nežádoucí účinky jako zimnice, bolesti hlavy, závratě, horečka, zvracení, alergické reakce, nauzea, bolest kloubů, pokles krevního tlaku či mírná bolest v dolní části zad, ale i jiné.
Nežádoucí účinky hlášené z klinických studií a z postmarketingových zkušeností:
Časté: Bolest hlavy, zrudnutí, nauzea, zvracení, únava, zimnice, pyrexie.
Méně časté: Chřipka, úzkost, vzrušení, letargie, rozmazané vidění, palpitace, výkyvy krevního tlaku, dyspnoe, epistaxe, průjem, stomatitida, bolest v horní části břicha, abdominální diskomfort, pruritus, kopřivka, studený pot, hyperhidróza, bolest zad, svalová křeč, bolest končetiny, bolest na hrudi, malátnost, bolest, nevolnost na hrudi, pocit abnormality, pocit chladu, pocit horka, onemocnění podobné chřipce, erytém v místě infuze, extravazace v místě infuze, bolest v místě infuze, anorexie.
Uchovávání: Uchovávejte při teplotě do 25 °C. Chraňte před mrazem, lahvička s rozpouštědlem by mohla prasknout. Uchovávejte v krabičce, aby byl přípravek chráněn před světlem. Nepoužívejte po uplynutí doby použitelnosti. Uchovávejte mimo dohled a dosah dětí.
Držitel rozhodnutí o registraci: Baxalta innovations GmbH, Industriestrasse 67, A-1221 Vídeň, Rakousko.
Registrační číslo: 75/152/00-C
Poslední revize SPC: 02.12.2020
*Všimněte si, prosím, změn v informacích o léčivém přípravku
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.