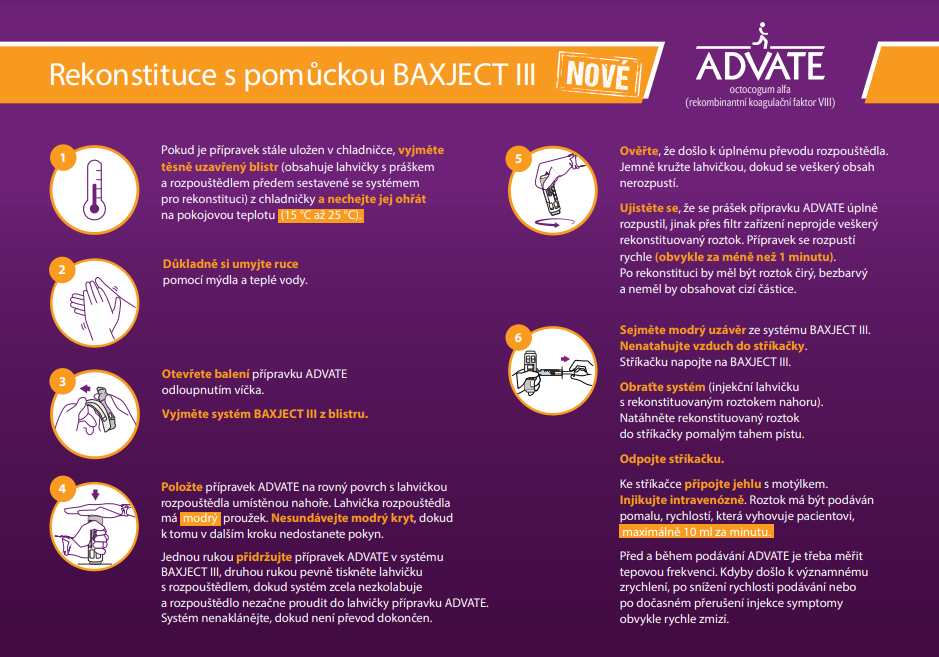
Zkrácené informace o léčivém přípravku
ADVATE 250 IU/5 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 500 IU/5 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 1 000 IU/5 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 1 500 IU/5 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 2 000 IU/5 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 3 000 IU/5 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 250 IU/2 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 500 IU/2 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 1 000 IU/2 ml prášek a rozpouštědlo pro injekční roztok
ADVATE 1 500 IU/2 ml prášek a rozpouštědlo pro injekční roztok
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Léčivá látka: Prášek: 250/500/1000/1500/2000/3000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 5 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 50/100/200/300/400/600 IU/ml lidského koagulačního faktoru VIII(rDNA) oktokogu alfa.
Prášek 250/500/1000/1500 IU lidského koagulačního faktoru VIII (rDNA), oktokogu alfa, Rozpouštědlo 2 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 125/250/500/750 IU/ml lidského koagulačního faktoru VIII (rDNA), oktokogu alfa.
Indikace: Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
Dávkování a způsob podání: Léčba on demand (dle potřeby): Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Dávka se určuje podle následujícího vzorce: Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5. Profylaxe:Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně. Způsob podání: ADVATE má být podáván intravenózně. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. Známé alergické reakce na myší nebo křeččí proteiny. Upozornění: Hypersenzitivita: U přípravku ADVATE byly hlášeny hypersenzitivní reakce alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Inhibitory: Tvorba neutralizujících protilátek (inhibitorů) faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Riziko vzniku inhibitorů souvisí se závažností onemocnění i s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 50 dnů expozice, ale pokračuje po celý život, přestože toto riziko není časté. Obecně platí, že všichni pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.Významné interakce: Nebyly hlášeny žádné interakce přípravků s lidským koagulačním faktorem VIII (rDNA) s jinými léčivými přípravky. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly. Hlavní nežádoucí účinky: Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší frekvencí, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka. Alergické reakce nebo hypersenzitivní reakce byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku). Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími hypersenzitivními reakcemi. K rozvoji neutralizujících protilátek (inhibitorů) může dojít u pacientů s hemofilií A, kteří jsou léčeni faktorem VIII, včetně přípravku ADVATE. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
Uchovávání: Uchovávejte v chladničce (2°C – 8°C). Chraňte před mrazem. Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po jedno období nepřesahující 6 měsíců. Přípravek nesmí být vrácen zpět do chladničky.
Držitel rozhodnutí o registraci: Takeda Manufacturing Austria AG, Industriestrasse 67, A-1221 Vídeň, Rakousko.
Registrační čísla: EU/1/03/271/001 - EU/1/03/271/020
Poslední revize SPC: 05/2025
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.