



Multidisciplinární péče o pacienty se syndromem krátkého střeva: proč ji centralizovat
MUDr. Anna Ouřadová, Fakultní nemocnice Královské Vinohrady, Interní klinika, Gastrocentrum
Článek
UEGW 2025: Přesnější cílení léčby ulcerózní kolitidy a „disease clearance“ jako dosažitelný cíl
MUDr. Jakub Chudý
Článek
ADVANCED CARE THROUGH GUT HEALING IN CROHN’S DISEASE
UEGW 2025 Berlin – satelitní sympozium firmy Takeda
MUDr. Kristýna Kubíčková,Ph.D. , ISCARE a.s., gastroenterologie
Článek
VDZ-CDST kalkulátor pro pacienty s CD
Nástroj, který pomáhá při rozhodování o další léčbě pacientů s CD
Článek
Predikce dlouhodobé odpovědi na léčbu pomocí aplikace CDST – ověření v klinické praxi
MUDr. Martin Lukáš, FASGEMUDr. Martin Lukáš, FASGE
Kazuistika
Dlouhodobá efektivní monoterapie vedolizumabem Crohnovské kolitidy
MUDr. Tomáš Douda, Ph. D , 2. interní GE klinika FN Hradec Králové
Kazuistika
Kazuistiky
Efektivní léčba těžké Crohnovské kolitidy ve 4. linii léčby monoterapií vedolizumabem
MUDr. Tomáš Douda, Ph. D, 2. interní gastroenterologická klinika FNHK
Kazuistika
Účinnost a bezpečnost vedolizumabu u starší pacientky s těžkou ulcerózní proktitidou
MUDr. Michal Stodola
Fakultní nemocnice Motol a Homolka, pracoviště Homolka, gastroenterologie
Kazuistika
Komplikovaná forma Crohnovy choroby s ileo-vezikálními píštělemi a sekundárním selháním anti-TNF terapie: efektivita vedolizumabu predikovaná CDST skóre
MUDr. Marek Veinfurt
interní oddělení a gastroenterologická ambulance, Karlovarská krajská nemocnice a.s., Nemocnice Cheb
Kazuistika
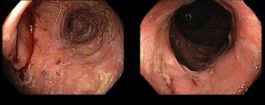
Pacient s Crohnovou chorobou a syndromem krátkého střeva, úspěšně léčený GLP-2 analogem teduglutidem (Revestive®)
MUDr. Jiří Vejmelka, MBA,
Interní klinika 3. LF UK a FTN Praha
Kazuistika
Užití vedolizumabu jako první linie léčby u pacienta s Crohnovou chorobou a komplikovanou akutní divertikulitidou
MUDr. Lukáš Farhád Mirchi, Ph.D.
Gastroenterologické oddělení a Interní klinika FNB a 3.LF UK
Kazuistika
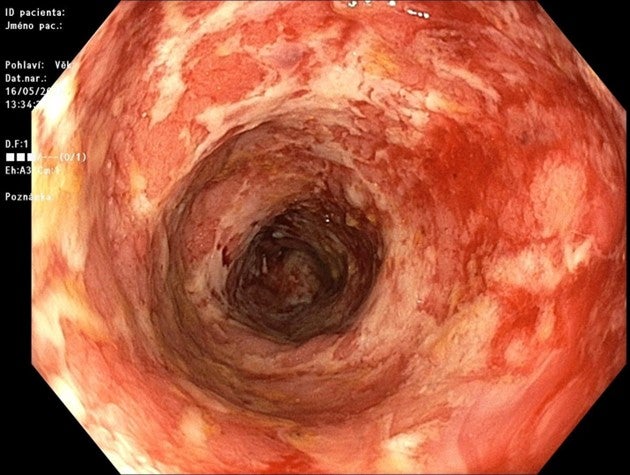
Vedolizumab, selektivní biologická léčba IBD
Za normálních okolností se na na některých naivních T a B lymfocytech (vč. bb přirozeného imun.systému), exprimuje epitop α4β7 ( transmembranózní adhezivní protein). Jeho ligandou je adhezivní molekula MAdCAM-1 (mucosal vascular addressin cell adhesion molecule 1), která se primárně objevuje na endoteliálních buňkách v GIT a GIT- asociovaných lymfatických tkáních. Díky jejich spojení dochází ke zpomalení valivého pohybu lymfocytů, až k možnosti průchodu do lamina propria.
Článek
Proč patří Entyvio® do předních linií? (CD)
Protože může dosáhnout i transmurálního zhojení.
Článek